


Farmaatsia või ebaseadusliku volituse kohta arutab Sasha Latypova oma eelmise aasta detsembri artiklis selle üle, kas EAÜ vastumeetmed on uuritavad ja eksperimentaalsed!
„Kohtunik, ma olin sunnitud võtma eksperimentaalse vaktsiini!“
Kaiser Permanente CA-s kaevatakse kohtusse:


Teine näide, mis pärineb juhtumist Roberts vs. Shriners. Kohtuasjas viidatakse õigele seadusele, mis kinnitab EUA-d (360bbb):



Seejärel väidab, et „FDA määratleb kõnealused ravimid uuritavatena“:

Seda ütleb FDA nende ravimite kohta avalikkusele, see ei ole see, kuidas neid seaduses määratletakse. Samuti FDA ütleb, et nad on nüüd „täielikult heaks kiidetud“, tegemist näib olevat EUA ainete õigusliku välistusega. FDA ütleb, et need tooted on „ohutud ja tõhusad“, kuigi ei ole selle väite põhistamist vastavalt tema valduses olevale tootjate esitatud dokumentatsioonile näidanud.
EAÜ ravim PHE ja praeguste PREP-seaduse deklaratsioonide kohaselt ei ole: uuritav ravim või eksperimentaalne. Vastavat staatust ei ole EAÜ-na seadusandja ette näinud.
Katherine Watt Bailiwicki uudistes: Bridges vs. Metodisti juhtum
Pange tähele, et Bridges vs. Metodisti juhtum lükati tagasi 12. juuni 2021 korralduses USDJ Lynn N. Hughesi poolt.
- aprillil 2021 teatas USA Texase osariigis asuv Houstoni metodisti haigla oma personalipoliitikast, mis nõuab kaitsepookimist COVID-19 vastu 7. juuniks, alustades juhtkonnast ja seejärel inokuleerides ülejäänud töötajad, kõik selle kulul.
Jennifer Bridges ja veel 116 isikut kaebasid COVID-19 vastu kaitsepookimise nõude töökaotuse ähvardusel ja töösuhte lõpetamise blokeerimiseks kohtusse. Kaebus väidab, et metodistide haigla sunnib ebaseaduslikult oma töötajaid süstima ühte praegu kättesaadavatest vaktsiinidest või vallandama. Haigla on asunud seda juhtumit lõpetama.
Bridges pühendab suurema osa oma väidetest väitele, et praegu kättesaadavad COVID-19 vaktsiinid on eksperimentaalsed ja ohtlikud. See väide on vale ja see on ka ebaoluline.
[Märkus: eetika loogilist arutluskäiku järgides võib jõuda teistsugusele järeldusele. Tegemist on kahe erineva ainedistsipliiniga, st praktiline õigus ja eetikateadus, mis võivad käeoleva juhtumi puhul eksisteerida erinevates teoreetilistes maailmades. – toim.]
Bridges väidab, et kui ta vallandatakse vaktsiini süstimisest keeldumise eest, lõpetatakse tema töösuhe ebaseaduslikult. Selle küsimuse lahendamisel ei võeta arvesse vaktsiinide ohutust ja efektiivsust.
Kohtunik Hughes kuulutas Bridgesi väite, et “vaktsiinid“ on eksperimentaalsed ja ohtlikud, „valeks“. Kohtunik näib olevat eksperimentaalse osa suhtes tehniliselt korrektne. Pange tähele, et „ohtlikku“ osa ei lubatud kunagi kohtus vaidlustada.
Seega kohtu hinnangul ei ole EUA vaktsiinid rangelt võttes meditsiinilised katsed.
Vaatame, mida tähendab „EUA“ regulatiivne staatus. Kas EAÜ vastumeetmed peavad järgima tavapärast head tootmistava? Vaatame USA koodeksit:


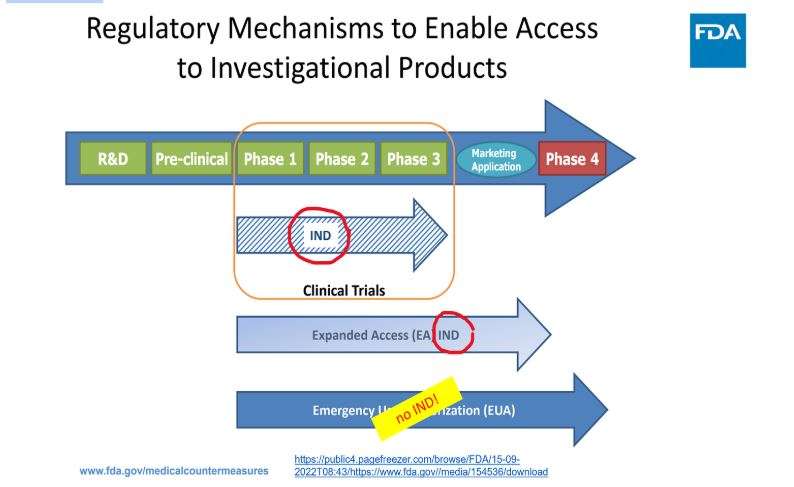
Järgneval slaidil on esitatud see, mida FDA avaldab oma „õigusliku valmisoleku“, st FD&C seadusest mööda navigeerimise kohta, PowerPointi esitlustes:


Teatud kaubanduslikele toodetele loa andmisel on kehtestatud erandid, mis võimaldavad tavapärastest loa väljastamise tõkenditest kõrvale navigeerida. Nimetagem seda EUA-ks. Sellel lehel on selgelt öeldud, et „EAÜ kasutamine ei ole uuritav, seega ei ole IRB heakskiit ja teadlik nõusolek vajalikud“.


Kahtluste vältimiseks: kliinilisi uuringuid (nagu seaduslikult määratletud inimkatsed) EAÜ puhul ei ole esitatud:

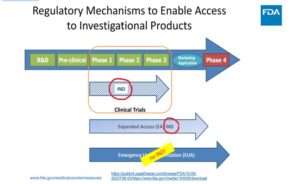
FDA on esitanud tõlgenduse, kuidas FD&C seadusest kõrvale navigeerida, luues selles eraldi jaotise (juhuslik arv 564) ja moodustades uue „regulatiivse“ tee, mis asub sisuliselt väljaspool tavapäraseid ravimieeskirju: „NO investigational review board, NO informed consent ja NO cGMP compliance“ (EI PUUDUB uurimiskomisjoni, EI OLE teadlikku nõusolekut ja EI OLE cGMP vastavust) kehtivad asjadele, mida nimetatakse „EUA vastumeetmed rahvatervise hädaolukorras“.
Kohtunik Hughesi hinnangul meditsiinilised katsed ehk „kliinilised uuringud“ on DFCA-s määratletud järgmiselt:
Kliiniline uuring tähendab mis tahes eksperimenti, mille käigus ravimit manustatakse, väljastatakse või kasutatakse ühe või mitme inimesega. Käesolevas osas tähendab eksperiment ravimi mis tahes kasutamist, välja arvatud turustatud ravimi kasutamine meditsiinipraktika käigus.
Kuid teine USA seadus eemaldab EUAd FDCA (ja FDA eeskirjade) reguleerimisalast, luues spetsiaalse „mitteuuritava“ klassi:
21 USC 360bbb-3(k): kui tootele on antud luba käesoleva jaotise alusel, ei käsitata sellise toote kasutamist loa kohaldamisalas kliinilise uuringuna käesoleva jaotise paragrahvi 355 punkti i, paragrahvi 360b punkti j või punkti 360j alapunkti g või käesoleva peatüki või rahvatervise seaduse [42 U.S.C. 262] paragrahvi 351 tähenduses.
Seetõttu ei ole meditsiinilised eksperimendid EUA-le võimalikud nende „mitte-uuritava“ staatuse tõttu. Kui kaubanduslikku toodet ei saa uurida, puudub menetlus ohutust, tõhusust ja tootmiskontrolli käsitlevate regulatiivsete tõendite kogumiseks, et tagada vastavus rahvatervise seaduse (PHS Act) paragrahvi 351 punktile a, (42 U.S.C. 262) ja cGMP-le (FD&C Acti paragrahvi 501 lõike a punkti 2 alapunkt B) (21 U.S.C. 351(a)(2)( B)) ja 21 CFR-i osale 210, 211 ja 610).
Elimineerimisprotsessi abil jõuame selleni:

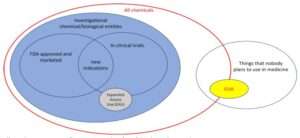
COVID-19 vastu kaitsepookimiseks kasutatavad süstitavad biotooted on kemikaalid ja bioloogilised ained, mis ei sobi ravimiks ja keegi ei kavatse neid sellisena kasutada. Vähemalt USA kehtivate seaduste alusel ehk juriidilise puuri tähenduses nende toodete nimetamine eksperimentaalseteks, uuritavateks, meditsiinilisteks või farmaatsiatoodeteks, puudub sellel väitel seaduslik alus, mis lubaks teha vastupidiseid vaidlustavatuid järeldusi. Need ei ole kumbki neist asjadest. Tegemist on toodetega, mis on toodud täitevvõimu esindajate heakskiidul turule, mida turustatakse riiklikult ja rahvusvahelises kaubanduses nende töövõtjate vahendusel.
Kohtunik Hughes on täpne, kui ta väidab, et COVID-19 biotoodete nimetamine eksperimentaalseks ja ohtlikuks on „vale ja ebaoluline“, sest EAÜ kohaldamise korral puudub alus, mis lubaks teha vääramatu järelduse, et need on juriidilises tähenduses eksperimentaalsed. Ohutus ei ole oluline EAÜ puhul, mis väljastatakse arvamuste põhjal ja mida ei kiideta heaks andmete põhjal. Kliiniliste uuringute andmeid ei ole vastavalt tavapärasele kliinilisele uuringule võimalik koguda ja võrrelda randomiseeritud metodoloogia alusel nii, et oleks võimalik hinnata riski-kasu, nagu on määratletud FDCA-s.
Kokkuvõtteks
Need on andmed, millele FDA tugines, et väljastada bioloogilise litsentsi kinnitus (BLA) Pfizeri algse “Wuhani variandi“ COVID-19 mod-RNA biotoote jaoks. See kaadrite versioon on sellest ajast alates turult eemaldatud ja asendatud uute versioonidega ilma kliiniliste uuringuteta.
BLA väljastamine on vaidlustatud väitega, et „EUA vastumeede” regulatiivne rada ei ole uurimisega seotud, mistõttu on võimatu läbi viia tavapäraseid kliinilisi uuringuid, st seadusega kaitstud inimkatseid. EUA vastumeetmete väljastamise vaidlustamisel on kokkuvõtteks tuginetud kahele väitele:
esiteks: EUA vastumeetmete ohutust või tõhusust ei saa testida vastavalt Ühendriikide seadusele (21 CFR 312 ja 21 CFR 601) ega vastavust kehtivatele headele tootmistavadele (cGMP) ega headele levitamistavadele (GxP üldiselt). Seega jõustab FDA seadusega ettenähtud EUA toodete „mitteuurimislik“ olemus. Mitte-uuringustaatus välistab kliinilise uuringu (inimkatse) võimaluse;
teiseks: EUA vastumeetmed ei ole rangelt võttes uuritavad ega selles tähenduses eksperimentaalsed, mille kohta leiate föderaalkohtu arutluskäigu siit.
Eelolev vaidlustuse alus võib olla relevantne ka Eesti siseturu puhul. Näiteks FDA tunnustab edaspidi Eesti Ravimiameti inspektsiooni tulemusi, nagu teatatakse 30. novembril 2018.
Ravimiamet kinnitab oma pressiteates:
FDA on … „jõudnud veendumusele, et Ravimiametil on olemas protseduurid ja piisavalt võimekust viimaks läbi GMP* inspektsioone inimtervishoius kasutatavate ravimite tootjate juures sellisel tasemel, et need vastavad USA Toidu ja Ravimiameti nõuetele“ [rõhuasetus lisatud].
Toimetas ja koostas Revo Jaansoo (14.12.2024)
Allikas: Sasha Latypova – Substack.com, 03.12.2023, Ravimiameti koduleht, 30.11.2018
⃰ Ravimite tootmise head tootmistavad (Good Manufacturing Practice, GMP) hõlmab üldpõhimõtteid ja kriteeriume, mida kasutatakse ravimitootmise käigushoidmiseks ja hindamiseks ülemaailmselt.






